原文链接:(http://www.nature.com/articles/s41524-017-0007-1)
Computational modeling sheds light on structural evolution in metallic glasses and supercooled liquids
Jun Ding & En Ma
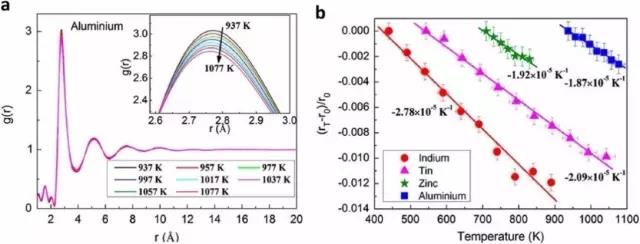
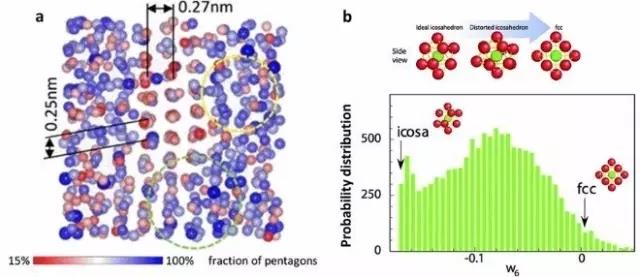
This article presents an overview of three challenging issues that are currently being debated in the community researching on the evolution of amorphous structures in metallic glasses and their parent supercooled liquids. Our emphasis is on the valuable insights acquired in recent computational analyses that have supplemented experimental investigations. The first idea is to use the local structural order developed, and in particular its evolution during undercooling, as a signature indicator to rationalize the experimentally observed temperature-dependence of viscosity, hence suggesting a possible structural origin of liquid fragility. The second issue concerns with the claim that the average nearest-neighbor distance in metallic melts contracts rather than expands upon heating, concurrent with a reduced coordination number. This postulate is, however, based on the shift of the first peak maximum in the pair distribution function and an average bond length determined from nearest neighbors designated using a distance cutoff. These can instead be a result of increasing skewness of the broad first peak, upon thermally exacerbated asymmetric distribution of neighboring atoms activated to shorter and longer distances under the anharmonic interatomic interaction potential. The third topic deals with crystal-like peak positions in the pair distribution function of metallic glasses. These peak locations can be explained using various connection schemes of coordination polyhedra, and found to be present already in high-temperature liquids without hidden crystal order. We also present an outlook to invite more in-depth computational research to fully settle these issues in future, and to establish more robust structure-property relations in amorphous alloys.